Proteins
can be analysed in a multitude of ways using a plethora of techniques. When it
comes to protein purification, there are certain pieces of information about
your protein that you are always interested in collecting. Any other aspects
you may need to measure are decided by the nature of your research project.
For
protein purification, the key pieces of information are identity, purity, size
homogeneity, activity and concentration of your target protein (it is also
worth trying to get some information about any key impurities as well).
Determining protein purity
Without doubt, the most common
technique for determining protein purity is denaturing gel electrophoresis by sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE).
This technique separates proteins by size and allows various detection
techniques to be used. Classic methods include Coomassie and silver staining,
but more and more pre-labeling using a fluorescent dye is gaining in
popularity. The classic staining methods use photographic detection using CCD
digital imagers (or film if you're lab is old school :) which
are robust but less sensitive and quantitative compared to fluorescent pre-labelling.
Alternatives
to SDS-PAGE for purity analysis include 2-D PAGE, size exclusion
chromatography, and mass spectrometry (Matrix-assisted laser
desorption/ionization; MALDI-MS).
Measuring
protein identity
The
most common technique used for protein identity is western blotting. Western
blotting uses denaturing SDS-PAGE gel electrophoresis followed by transfer of
the separated proteins to a membrane. These proteins are then detected by a
specific antibody and a secondary functionalized antibody which enables
detection by chemiluminescence or fluorescence.
Mass
spectrometry in combination with reverse phase chromatography, can provide an
easy and fast complement or alternative to the antibody-based detection step in
western blotting. The main drawback is that you need to have access to a mass
spectrometer, a significant piece of kit and often a shared service. The main
principle for confirming protein identity using mass-spectrometry is to
trypsinate the SDS-PAGE gel band of interest prior to mass analysis. To
identify the peptides, Electrospray Ionization (ESI) connected on-line with
reverse phase chromatography is common. The mass-spectrum of peptide species
after trypsinization provides a unique fingerprint for most proteins, which can
be identified using a database lookup.
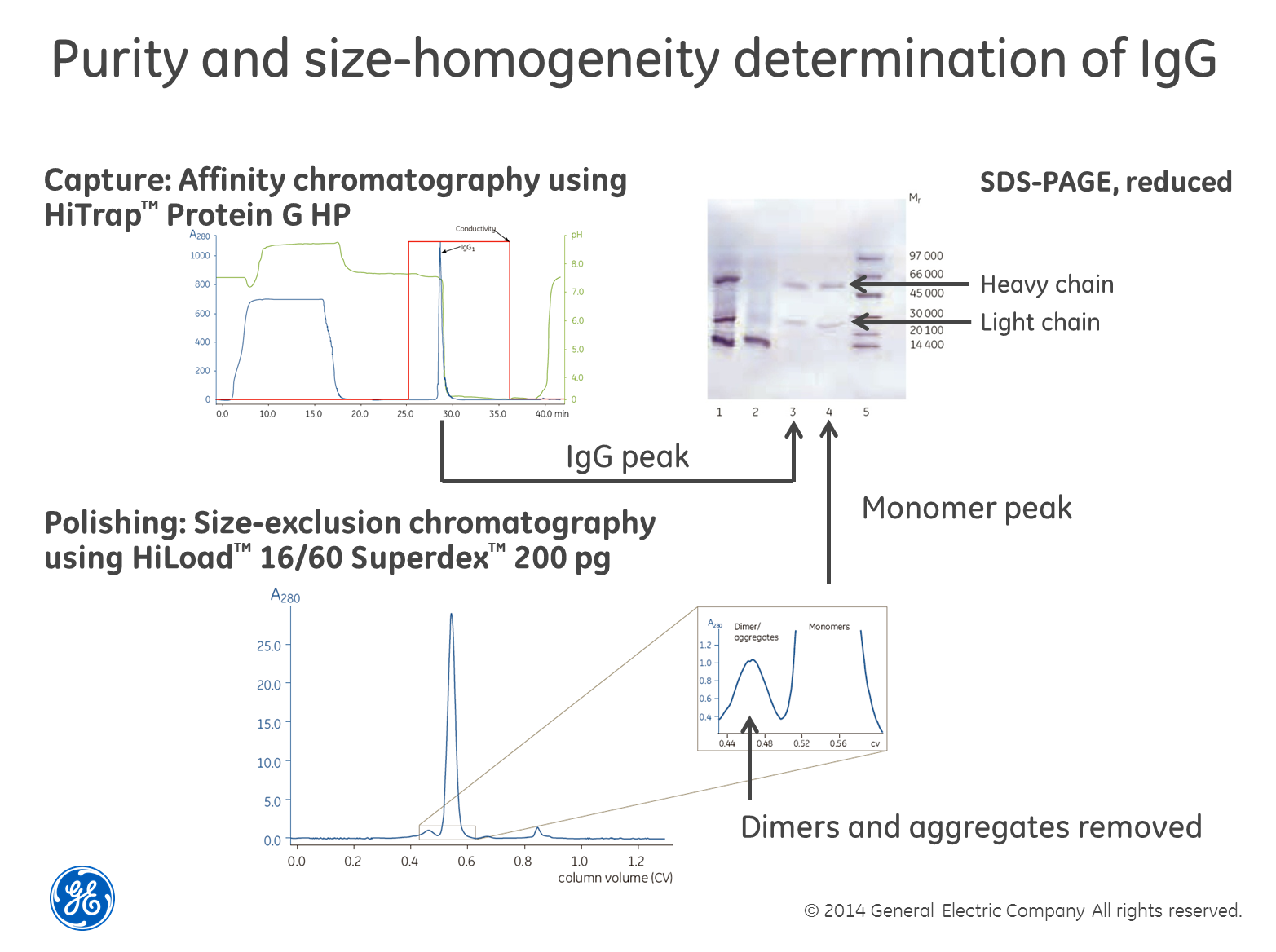
Use
SEC in combination with SDS-PAGE for size homogeneity
Perhaps
the most robust and powerful way of determining size homogeneity is to first
separate your sample using size-exclusion chromatography (SEC), collect the
fractions and then run a SDS-PAGE gel containing reducing agents such as dithiothreitol
(DTT) on the fractions. The reducing agent breaks di-sulfide
bridges between cysteine residues and the gel shows single-chain sub-units of
the different sizes, if cysteins are causing multimerization. The textbook
example is the combined purification and analysis of IgG as
exemplified in the image above. The only drawback we can think of with this
method is that if you have a low concentration of the protein in your sample,
it may be difficult to detect. You also need to choose a SEC column with the
right separation range for your protein of course.
Alternatives
to this approach include using light-scattering or mass-spectrometry after the
SEC step. Again, these detection techniques involve investment in expensive
instruments.
Estimating
concentration
As
the subhead suggests, we think regardless of what measurement technique you
use, you are likely to end up with nothing more than an estimation of
the concentration. That said, measuring concentration is a chapter of its own
(something we’re going to discuss in detail in future posts) but unfortunately
no protein concentration assay method exists that is either specific to
proteins or uniformly sensitive to all protein types (i.e. not affected by
differences in protein composition). It is therefore important to choose the
method that is most compatible with the sample and will give enough information
for you to move forward with you research.
For example, one of the most common
methods when you want to check the expression level of you target protein from
cultivation is to do a rough estimation with SDS-PAGE; it will show if you are
on track. If you want to measure the total protein concentration the tried and
tested methods are the Coomassie (Bradford) protein assay, BCA protein assay
(also known as Smith assay) and UV absorbance at 280nm.
Each of these methods has its own
set of advantages and disadvantages. However, these methods give only an
estimation of the total protein concentration. Because no method can be
considered the ideal assay method for all circumstances, most researchers have
more than one type of protein assay available in their laboratories. The
BCA Protein Assay and Bradford Protein Assay methods are complementary and
cover most samples, with both based on detection of color change. BCA is a
two-step protocol including a Protein-copper chelation and secondary detection
of the reduced copper. Bradford is a protein-dye binding and direct detection
of the color change associated with the bound dye.
When
choosing an assay somethings to consider:
•Compatibility
with the sample type and components (e.g. in lysis buffer) that may interfere
with the protein and/or the reagents in the assay used
•The concentration range of the
assay and required sample volume. For example the Bradford assay works in a
concentration range of 125–1,000 μg BSA /ml and the BCA assay in a working
range of 20-2000μg BSA/ml
•
Protein compositional differences which end up in different amount of color in
the final solution and may give wrong concentrations- choose assay and protein
standard which will minimise this error
•Speed
and convenience for the number of samples to be tested
•Availability
of spectrophotometer or plate reader
If
your protein is an enzyme with activity in a specific enzymatic assay, using an
assay may be an easy way to find out where in your eluted purification
fractions the target protein is. You will then be able to detect your protein
through all purification steps and have full control of the design of the
purification protocol and quality of the obtained preparation. The activity is
also an insurance that the protein is obtained in its native state.
We
will go through the methods described in upcoming posts when we delve into our
DHFR project.
In
the meantime, thanks for reading and if you have any questions let us know via
the comments section below.